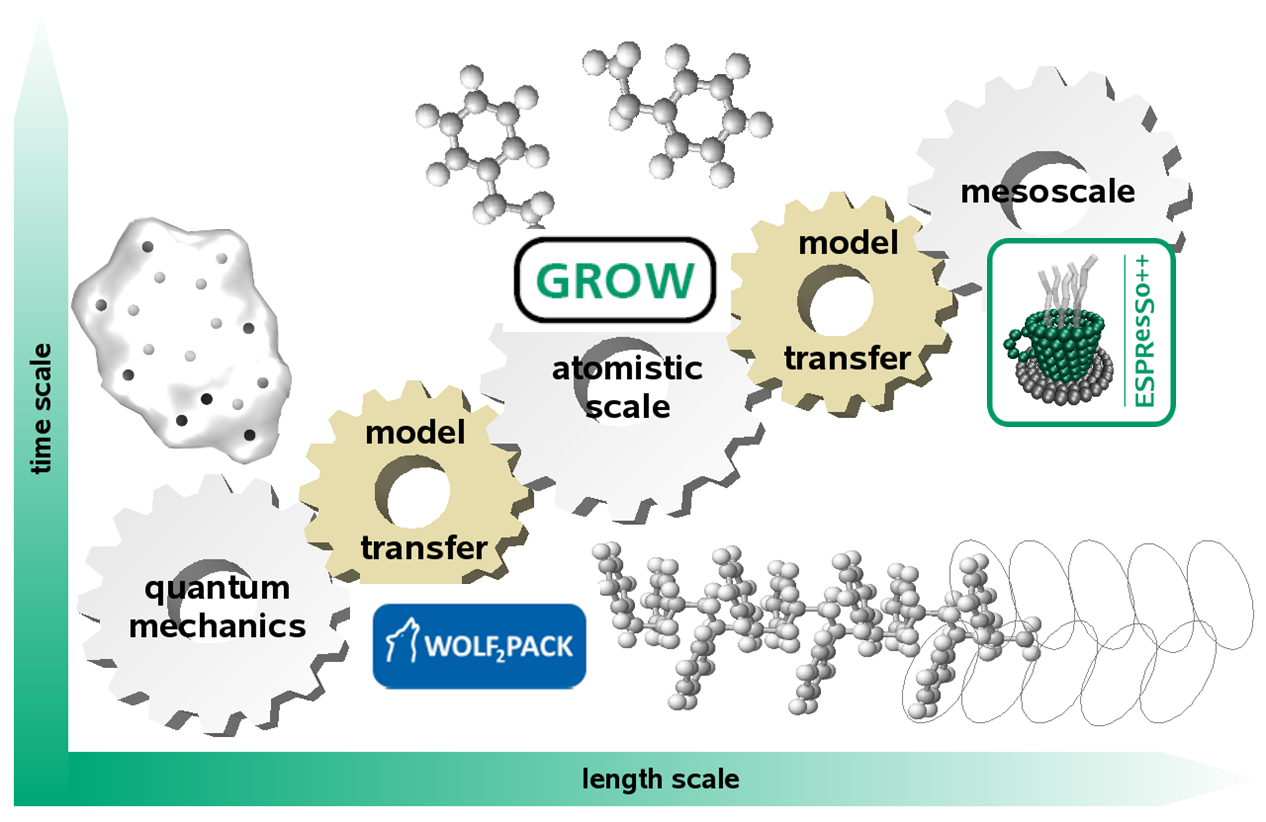
Ein grundlegendes Ziel der computergestützten Chemie – seien es Newton'sche oder quantenmechanischen Verfahren - ist die akkurate Modellierung von physikalischen Kräften und Energien. Durch zuverlässiges Modellieren der zugrunde liegenden Kräfte bieten molekulare Simulationen atomistische Einblicke in makroskopische Versuchsbeobachtungen.
Obwohl es einige kommerzielle Entwicklungen gibt, stammt die hierzu verwendete Software eher von Wissenschaftlern als von IT-Spezialisten. Grund dafür sind die anspruchsvollen und sich fortlaufend entwickelnden Konzepte der Physik und Chemie, die in geeignete algorithmische Lösungen umgewandelt werden müssen. Wir verfolgen den Grundansatz, die Software wachsen zu lassen, durch eine konsequente Modularisierung der darin befindlichen Funktionalität. Auf diese Weise können algorithmische Lösungen in einer gekapselten Form eingesetzt werden, sodass wir bei Bedarf bestimmte Funktionalitäten leicht identifizieren und erweitern können. Basierend auf dieser Idee hat unsere Gruppe verschiedene unabhängige Software-Tools entwickelt.