Deep Learning für Virtual Material Design
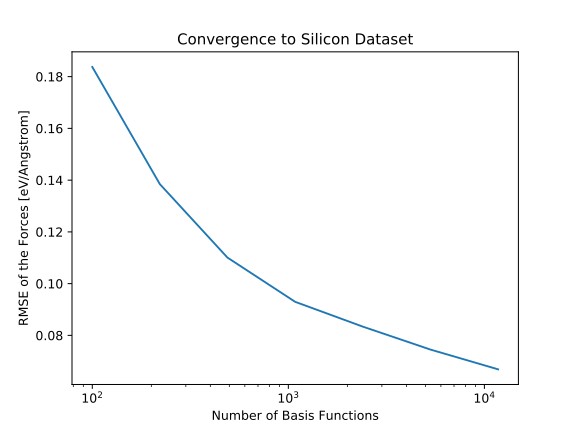
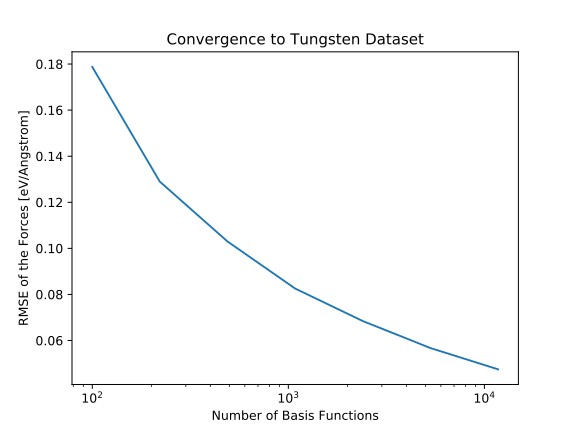
Empirische Analysepotenziale und ab initio-Methoden wie die Dichtefunktionstheorie sind klassische Säulen der computergestützten Materialwissenschaft. Mit den Fortschritten im Bereich des maschinellen Lernens und der rasanten Zunahme der Rechenleistung werden datenbasierte Ansätze zu einem neuen Werkzeug, mit dem die Vorhersagekraft von ab initio-Methoden mit der rechnerische Effizienz von empirischen Potenzialen kombiniert werden können.
Standard-Techniken des maschinellen Lernens wie Kernel-Learning (z.B. für das Gaußsche Approximationspotenzial), tiefe neuronale Netze (z.B. neuronale Netzwerkpotenziale von Behler et al.) und verallgemeinerte lineare Modelle (z.B. für Impuls-Tensor-Potentiale) wurden verwendet, um aus Daten schnelle und genaue Kraftfelder zu entwickeln, ohne dass menschliches Wissen über die zugrunde liegende Chemie erforderlich war.
In diesem Projekt entwickeln wir qualitativ hochwertige, einfach zu handhabende Implementierungen solcher maschinellen Lernpotenziale und untersuchen Verbesserungsmöglichkeiten.