Deep Learning for Virtual Material Design
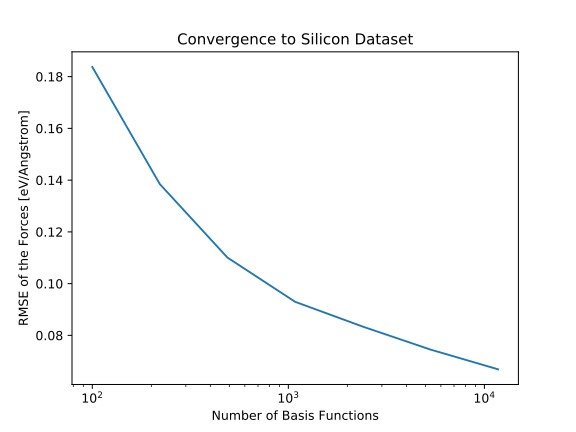
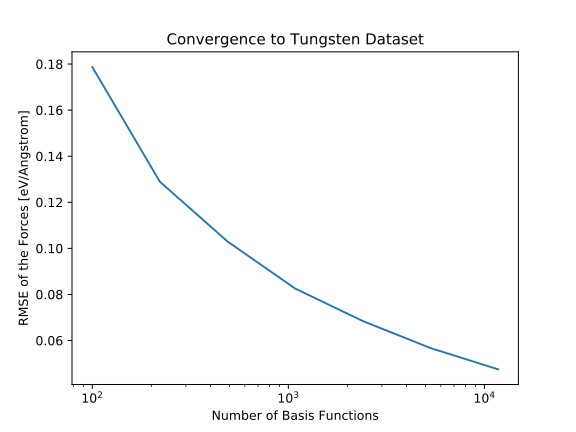
Empirical analysis potentials and ab-initio methods such as density function theory have been the pillars of computer-aided materials science. With theoretical advances in machine learning and the rapid increase in computing power, data-based approaches have emerged a new class of models with the goal of combining the predictive power of ab-initio methods and the computational efficiency of empirical potentials.
Standard machine learning techniques such as kernel learning (e.g. for the Gaussian approximation potential), deep neural networks (e.g. neural network potentials by Behler et al.), and generalized linear models (e.g. for momentum tensor potentials) have been employed to develop fast and accurate force fields from data without the need for human knowledge about the underlying chemistry.
In this project we develop high-quality, easy-to-use implementations of such machine learning potentials and investigate possibilities to improve the existing approaches by utilizing modern tools from the constantly growing toolbox of data science.